Types of congenital epidermolysis bullosa
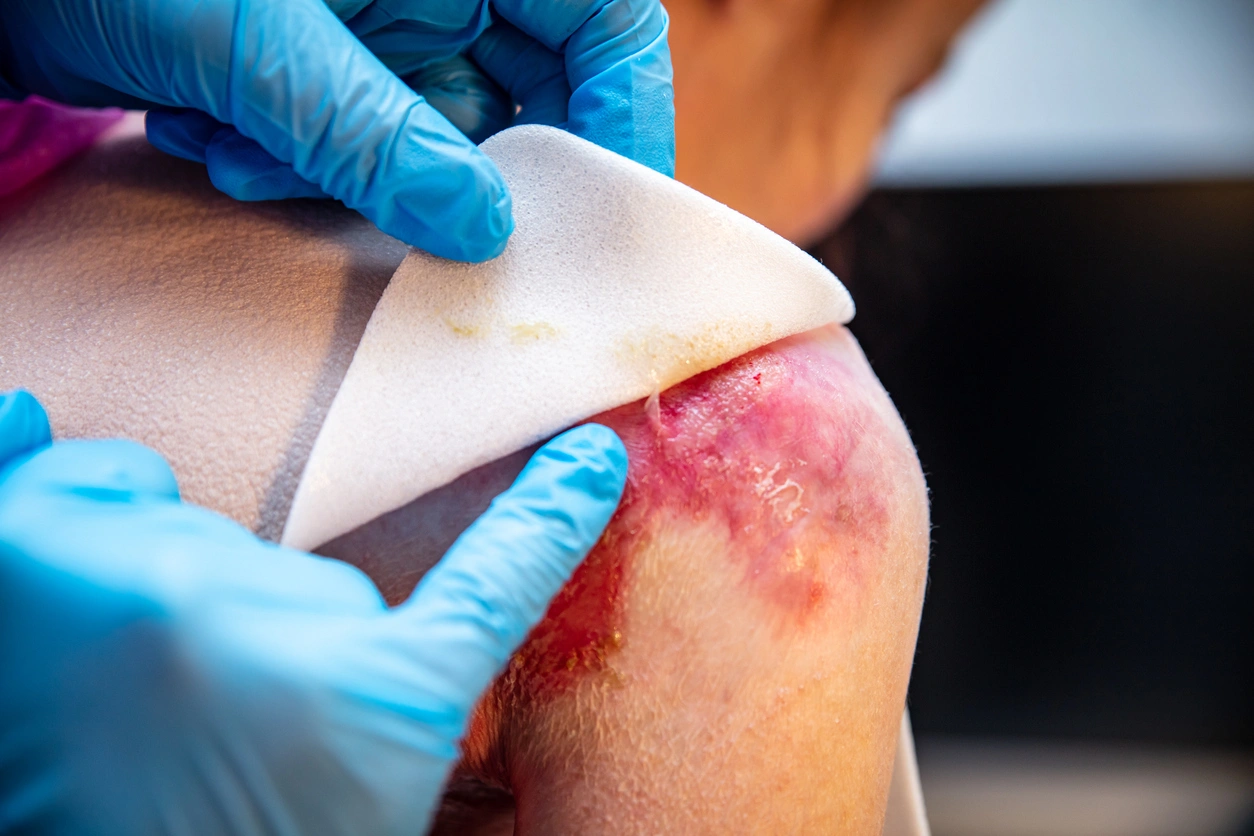
Patients with congenital epidermolysis bullosa (EB) suffer from highly fragile skin and/or mucous membranes, which blister with minimal friction or mechanical stress. Medical professionals classify EB into several types based on the layer of skin where blistering occurs and which proteins or genes are implicated. Understanding the types of congenital epidermolysis bullosa allows clinicians to predict the severity of the condition, plan treatment, and offer guidance to affected individuals and their families.
What are the different types of epidermolysis bullosa?
The classification of EB depends on which part of the skin structure is affected and the specific genetic mutations involved. This classification system divides EB into four main types, each with multiple subtypes characterized by varying degrees of severity, blistering patterns, and associated features. It is important to correctly identify these types and subtypes in order to determine a management plan and treatment of epidermolysis bullosa tailored to each patient.
Epidermolysis bullosa simplex (EBS)
Epidermolysis bullosa simplex (EBS) occurs when blistering happens within the epidermis, the outermost layer of skin. It is the most common form of inherited EB and often presents in infancy or early childhood. In EBS, defects in structural proteins that help support the cytoskeleton of skin cells make the epidermis more vulnerable to mechanical stress. As a result, blisters form in response to even minor friction or bumps.
The genetic mutations most commonly associated with EBS affect the keratin 5 (KRT5) and keratin 14 (KRT14) genes. These genes produce keratin proteins that maintain the structure and integrity of epidermal cells. When these proteins are dysfunctional, the basal layer of the epidermis cannot withstand the shear forces between cells, resulting in blister formation. EBS follows an autosomal dominant inheritance pattern, meaning that a single mutated gene from one parent is sufficient to cause the condition.
The severity of EBS varies. Some individuals experience mild blistering confined to the hands and feet, while others have more widespread blistering that can also affect mucous membranes. Typically, EBS does not cause scarring because the blistering occurs within the epidermis, rather than deeper layers.
Junctional epidermolysis bullosa (JEB)
Junctional epidermolysis bullosa (JEB) results from defects in proteins found at the lamina lucida, a layer in the basement membrane zone between the epidermis and dermis. JEB typically develops at birth or in early infancy and often follows autosomal recessive inheritance, meaning that an affected individual inherits two copies of the mutated gene—one from each parent.
Proteins involved in JEB play a critical role in anchoring the outer skin layer to the deeper tissues. Mutations in genes like LAMB3 and LAMC2, which code for laminin and collagen components, weaken this connection. As a result, blisters form at a deeper level within the skin, leading to more significant skin loss and complications involving mucous membranes, such as in the mouth, eyes, and gastrointestinal tract.
JEB can present in various forms, ranging from moderately severe to life-threatening conditions in infancy. Some subtypes also involve other body systems, including abnormalities in teeth and hair, highlighting the widespread impact of the proteins involved in JEB.
Dystrophic epidermolysis bullosa (DEB)
Dystrophic epidermolysis bullosa (DEB) affects the zone below the lamina densa in the upper dermis. It results from mutations in the COL7A1 gene, which encodes type VII collagen, a protein that forms anchoring fibrils to secure the epidermis to the dermis. When these fibrils are defective or absent, the skin layers separate under minimal pressure, causing blistering.
DEB can be inherited in either an autosomal dominant or autosomal recessive pattern, with recessive forms often being more severe. Individuals with DEB experience slow-healing blisters that lead to extensive scarring, which can cause the fusion of fingers and toes (known as pseudosyndactyly). Chronic wounds are common, and the risk of developing squamous cell carcinoma—a common type of non-melanoma skin cancer—in long-standing wounds increases over time.
There are several subtypes of DEB, with dominant forms generally causing milder symptoms, while recessive forms tend to result in more severe blistering and complications affecting mucous membranes. The degree of scarring and disability varies between individuals and across subtypes.
Kindler syndrome (KS)
Kindler syndrome (KS) is distinct from the other main types of EB because blistering can occur at multiple levels within the skin, making it more complex. KS is caused by mutations in the FERMT1 gene, which affects the production of kindlin-1, a protein involved in cell adhesion and the structural integrity of the skin.
The condition also evolves over time, with symptoms becoming more pronounced as the individual ages. In infancy and early childhood, individuals with Kindler syndrome typically have fragile skin that blisters easily. As they grow older, they develop sensitivity to sunlight (photosensitivity), changes in skin pigmentation, thinning, and atrophy of the skin tissue, along with other skin abnormalities. The condition can also affect mucosal tissues, including the mouth and gastrointestinal tract.
Since Kindler syndrome affects the skin’s response to UV exposure as well as mechanical stress, individuals with this condition require sun protection and careful management of friction injuries to prevent further skin damage.
References
- Has C, et al. Inherited epidermolysis bullosa: updated classification and recommendations. Br J Dermatol. 2020. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC3750879/
- Piel de Mariposa. Types of EB [Internet]. Available from: https://pieldemariposa.es/en/types-of-eb/
- MSD Manuals. Epidermolysis bullosa: Dermatologic disorders [Internet]. Available from: https://www.msdmanuals.com/professional/dermatologic-disorders/bullous-diseases/epidermolysis-bullo…;